Aturan Bent

Dalam kimia, Aturan Bent menggambarkan dan menjelaskan hubungan antara hibridisasi orbital dan keelektronegatifan substituen.[1][2] Aturan ini dinyatakan oleh Henry A. Bent sebagai berikut:[2]
Karakter s atom terkonsentrasi dalam orbital yang diarahkan menuju substituen elektropositif.
Teori ikatan valensi memberikan pendekatan yang baik terhadap struktur molekul. Aturan Bent mengatasi perbedaan antara geometri yang teramati dan yang diidealkan.[3] Menurut Aturan Bent, sebuah atom pusat yang berikatan dengan beberapa gugus akan mengalami rehibridisasi sehingga orbital dengan lebih banyak karakter s diarahkan menuju gugus elektropositif, dan orbital dengan lebih banyak karakter p diarahkan menuju gugus yang lebih elektronegatif. Dengan menghilangkan anggapan bahwa semua orbital hibrida adalah setara, Aturan Bent menghasilkan prediksi yang lebih baik tentang geometri molekul dan kekuatan ikatan.[4][5] Aturan Bent dapat dibenarkan melalui tingkat energi relatif dari orbital s dan p. Aturan Bent merepresentasikan modifikasi dari teori VSEPR untuk molekul dengan simetri yang lebih rendah dari ideal.[6] Untuk ikatan dengan atom yang lebih besar dari periode bawah, tren dalam hibridisasi orbital sangat bergantung pada baik keelektronegatifan maupun ukuran orbital.
Sejarah
Pada awal tahun 1930-an, tak lama setelah banyak perkembangan awal dalam mekanika kuantum, teori-teori tersebut mulai diterapkan pada struktur molekul oleh Pauling,[7] Slater,[8] Coulson,[9] dan lainnya. Secara khusus, Pauling memperkenalkan konsep hibridisasi, di mana orbital atom s dan p digabungkan untuk menghasilkan orbital hibrida sp, sp2, dan sp3. Orbital hibrida terbukti kuat dalam menjelaskan geometri molekul dari molekul sederhana seperti metana, yang memiliki bentuk tetrahedral dengan atom karbon sp3 dan sudut ikatan 109.5° antara empat ikatan C-H yang setara. Namun, penyimpangan kecil dari geometri ideal ini mulai tampak pada tahun 1940-an.[10] Salah satu contoh yang sangat terkenal adalah air, di mana sudut antara dua ikatan O-H hanya 104.5°. Untuk menjelaskan ketidaksesuaian tersebut, diusulkan bahwa hibridisasi dapat menghasilkan orbital dengan karakter s dan p yang tidak sama. A. D. Walsh pada tahun 1947[10] menggambarkan suatu hubungan antara keelektronegatifan gugus yang berikatan dengan karbon dan hibridisasi atom karbon tersebut. Akhirnya, pada tahun 1961, Bent menerbitkan tinjauan besar literatur yang mengaitkan struktur molekul, hibridisasi atom pusat, dan keelektronegatifan substituen[2] dan dari karya inilah nama Aturan Bent berasal.
Makalah asli Bent menganggap keelektronegatifan gugus dari gugus metil lebih kecil daripada atom hidrogen karena substitusi metil menurunkan konstanta disosiasi asam dari asam format dan dari asam asetat.[2]
Nonbonding orbitals
Aturan Bent dapat diperluas untuk merasionalkan hibridisasi dari orbital non-ikatan juga. Di satu sisi, sebuah pasangan elektron bebas (sebuah orbital non-ikatan terisi) dapat dianggap sebagai kasus batas dari substituen elektropositif, dengan kerapatan elektron sepenuhnya terpolarisasi menuju atom pusat. Aturan Bent memprediksi bahwa, untuk menstabilkan elektron non-ikatan yang tidak dibagi dan sangat terikat, orbitals pasangan bebas harus mengambil karakter s yang tinggi. Di sisi lain, sebuah orbital non-ikatan tak terisi (kosong) dapat dianggap sebagai kasus batas dari substituen elektronegatif, dengan kerapatan elektron sepenuhnya terpolarisasi menuju ligan dan menjauhi atom pusat. Aturan Bent memprediksi bahwa, untuk menyisakan sebanyak mungkin karakter s bagi orbital terisi yang tersisa, orbital non-ikatan tak terisi harus memaksimalkan karakter p.
Secara eksperimental, kesimpulan pertama sejalan dengan sudut ikatan yang berkurang pada molekul dengan pasangan bebas seperti air atau amonia dibandingkan dengan metana, sementara kesimpulan kedua sesuai dengan struktur planar molekul dengan orbital non-ikatan tak terisi, seperti borana monomer dan ion karbenium.
Konsekuensi
Aturan Bent dapat digunakan untuk menjelaskan tren dalam struktur molekul dan reaktivitas. Setelah menentukan bagaimana hibridisasi atom pusat memengaruhi suatu sifat tertentu, elektronegativitas substituen dapat diperiksa untuk melihat apakah aturan Bent berlaku.
Sudut ikatan: Teori VSEPR dan Aturan Bent
Teori ikatan valensi memprediksi bahwa metana berbentuk tetrahedral dan bahwa etilena berbentuk planar. Pada air dan amonia, situasinya lebih rumit karena sudut ikatannya masing-masing 104,5° dan 107°, yang lebih kecil daripada sudut tetrahedral yang diharapkan yaitu 109,5°. Salah satu penjelasan untuk penyimpangan tersebut adalah teori VSEPR, di mana elektron valensi diasumsikan berada dalam daerah terlokalisasi dan pasangan elektron bebas diasumsikan saling menolak lebih besar daripada pasangan ikatan. Aturan Bent memberikan penjelasan alternatif.

Teori tolakan pasangan elektron kulit valensi (VSEPR) memprediksi geometri molekul.[11][12] Teori VSEPR memprediksi geometri molekul mengambil konfigurasi yang memungkinkan pasangan elektron berjauhan sejauh mungkin.[11][12] Pemaksimalan jarak elektron ini terjadi untuk mencapai distribusi elektron yang paling stabil.[11][12] Hasil teori VSEPR adalah kemampuan memprediksi sudut ikatan dengan akurat. Menurut teori VSEPR, geometri sebuah molekul dapat diprediksi dengan menghitung berapa banyak pasangan elektron dan atom yang terhubung ke atom pusat.[11][12] Aturan Bent menyatakan "[K]arakter s atom terkonsentrasi dalam orbital yang diarahkan menuju substituen elektropositif".[2] Aturan Bent menyiratkan bahwa sudut ikatan akan menyimpang dari sudut yang diprediksi oleh teori VSEPR; elektronegativitas relatif atom-atom yang mengelilingi atom pusat akan memengaruhi geometri molekul.[5] Teori VSEPR menyarankan cara memprediksi bentuk molekul secara akurat menggunakan aturan sederhana.[13] Namun, teori VSEPR hanya memprediksi sudut ikatan yang teramati secara mendekati.[13][14] Di sisi lain, aturan Bent lebih akurat.[5] Selain itu, telah ditunjukkan bahwa aturan Bent mendukung perhitungan mekanika kuantum ketika menggambarkan geometri molekul.[15]
| Molecule | Bond angle between substituents | 3D images with bond angles |
|---|---|---|
 Dimethyl ether
|
111.5° ± 1.5°[16]
|
 |
 Methanol
|
108.5° ± 2°[17]
|
 |
 Water
|
104.5°[18]
|
 |
 Oxygen difluoride
|
104.2°[19]
|
 |
Tabel di atas menunjukkan perbedaan antara sudut ikatan yang diprediksi oleh teori VSEPR dan sudutnya di dunia nyata. Menurut teori VSEPR, dietil eter, metanol, air, dan difluorida oksigen seharusnya memiliki sudut ikatan sebesar 109,5o.[12] Dengan menggunakan teori VSEPR, semua molekul ini seharusnya memiliki sudut ikatan yang sama karena mereka memiliki bentuk “bengkok” yang sama.[12] Namun, jelas bahwa sudut ikatan antara semua molekul ini menyimpang dari geometri idealnya dengan cara yang berbeda. Aturan Bent dapat membantu menjelaskan ketidaksesuaian yang tampak ini.[5][20][21] Substituen elektronegatif akan memiliki lebih banyak karakter p.[5][20] Sudut ikatan memiliki hubungan proporsional dengan karakter s dan hubungan terbalik dengan karakter p.[5] Dengan demikian, ketika substituen menjadi lebih elektronegatif, sudut ikatan molekul harus menurun. Dimetil eter, metanol, air, dan difluorida oksigen mengikuti tren ini sebagaimana diharapkan (sebagaimana ditunjukkan pada tabel di atas). Dua gugus metil adalah substituen yang terikat pada oksigen pusat dalam dietil eter. Karena kedua gugus metil bersifat elektropositif, karakter s yang lebih besar akan diamati dan sudut ikatan nyata lebih besar daripada sudut ikatan ideal 109,5o. Metanol memiliki satu substituen metil elektropositif dan satu substituen hidrogen elektronegatif. Karena itu, karakter s yang diamati lebih sedikit dibandingkan dimetil eter. Ketika terdapat dua gugus substituen hidrogen, sudut semakin menurun karena peningkatan keelektronegatifan dan karakter p. Akhirnya, ketika kedua substituen hidrogen digantikan oleh fluor dalam difluorida oksigen, terjadi penurunan sudut ikatan yang lain. Fluor sangat elektronegatif, sehingga menyebabkan penurunan signifikan dalam sudut ikatan.
Dalam memprediksi sudut ikatan air, aturan Bent menyatakan bahwa orbital hibrida dengan lebih banyak karakter s harus diarahkan menuju pasangan elektron bebas, sehingga orbital dengan lebih banyak karakter p diarahkan menuju atom hidrogen, menghasilkan penyimpangan dari orbital hibrida O(sp3) teridealisasi dengan 25% karakter s dan 75% karakter p. Dalam kasus air, dengan sudut HOH 104,5°, orbital pengikatan OH dibentuk dari orbital O(~sp4.0) (~20% s, ~80% p), sementara pasangan elektron bebas terdiri dari orbitals O(~sp2.3) (~30% s, ~70% p). Seperti dibahas dalam justifikasi di atas, pasangan elektron bebas bertindak sebagai substituen yang sangat elektropositif dan memiliki kelebihan karakter s. Akibatnya, elektron pengikatan memiliki karakter p yang meningkat. Peningkatan karakter p dalam orbital tersebut menurunkan sudut ikatan di antara keduanya menjadi kurang dari 109,5° (sudut tetrahedral). Logika yang sama dapat diterapkan pada amonia (sudut ikatan HNH 107,0°, dengan tiga orbital pengikatan N(~sp3.4 atau 23% s) dan satu pasangan bebas N(~sp2.1 atau 32% s)), contoh kanonis lain dari fenomena ini.
Tren yang sama berlaku untuk senyawa yang mengandung nitrogen. Berlawanan dengan ekspektasi teori VSEPR namun konsisten dengan aturan Bent, sudut ikatan amonia (NH3) dan nitrogen trifluorida (NF3) masing-masing adalah 107° dan 102°.
Tidak seperti teori VSEPR, yang dasar teoretisnya kini tampak goyah, aturan Bent masih dianggap sebagai prinsip penting dalam kajian modern mengenai ikatan.[5][22] Misalnya, modifikasi dari analisis ini masih dapat diterapkan, bahkan jika pasangan elektron bebas pada H2O dianggap tidak ekuivalen berdasarkan simetrinya (yaitu, hanya orbital oksigen s, dan px serta py dalam bidang yang dihibdrisasi untuk membentuk dua orbital pengikatan O-H σO-H dan pasangan bebas nO(σ), sementara pz menjadi pasangan bebas nO(π) berkarakter p murni yang tidak ekuivalen), seperti pada kasus pasangan bebas dalam metode natural bond orbital.
Untuk molekul tetrahedral seperti difluorometana dengan dua jenis atom yang terikat pada atom pusat, ikatan C–F menuju substituen yang lebih elektronegatif (F) akan melibatkan orbital karbon dengan karakter s yang lebih sedikit dibandingkan ikatan C–H, sehingga sudut antara ikatan C–F lebih kecil daripada sudut tetrahedral 109,5°.[15][23]
Molekul trigonal bipiramida memiliki posisi aksial dan ekuatorial. Jika terdapat dua jenis substituen, substituen yang lebih elektronegatif akan lebih menyukai posisi aksial karena sudut ikatan antara substituen aksial dan substituen elektronegatif lebih kecil daripada antara dua substituen ekuatorial.[23]
Bond lengths
Serupa dengan sudut ikatan, hibridisasi suatu atom dapat dikaitkan dengan panjang ikatan yang dibentuknya.[2] Ketika orbital pengikatan meningkat dalam karakter s, panjang ikatan σ ikatan menurun.
| Molekul | Panjang rata-rata ikatan karbon–karbon |
|---|---|
 |
1,54 Å |
 |
1,50 Å |
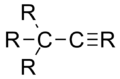 |
1,46 Å |
Dengan menambahkan substituen elektronegatif dan mengubah hibridisasi atom pusat, panjang ikatan dapat dimanipulasi. Jika suatu molekul mengandung struktur X-A--Y, penggantian substituen X dengan atom yang lebih elektronegatif mengubah hibridisasi atom pusat A dan memperpendek ikatan A--Y yang bersebelahan.
| Molekul | Panjang rata-rata ikatan karbon–fluor |
|---|---|
 Fluoromethane |
1,388 Å |
 Difluoromethane |
1,358 Å |
 Trifluoromethane |
1,329 Å |
 Tetrafluoromethane |
1,323 Å |
Ikatan antara unsur dengan keelektronegatifan yang berbeda akan bersifat polar dan kerapatan elektron dalam ikatan semacam itu akan bergeser ke arah unsur yang lebih elektronegatif. Menerapkan ide ini pada molekul fluoromethane menunjukkan kekuatan aturan Bent. Karena karbon lebih elektronegatif daripada hidrogen, kerapatan elektron dalam ikatan C–H akan dipendekkan dan ikatan C–F akan dipanjangkan.
Kecenderungan yang sama juga berlaku untuk analog terklorinasi dari metana, meskipun efeknya kurang dramatis karena klorin kurang elektronegatif dibandingkan fluor.[2]
| Molekul | Panjang rata-rata ikatan karbon–klorin |
|---|---|
 Chloromethane |
1,783 Å |
 Dichloromethane |
1,772 Å |
 Trichloromethane |
1,767 Å |
 Tetrachloromethane |
1,766 Å |
Kasus-kasus di atas tampaknya menunjukkan bahwa ukuran klorin kurang penting dibandingkan elektronegatifitasnya. Prediksi berdasarkan sterik saja akan menghasilkan kecenderungan sebaliknya, karena substituen klorin yang besar akan lebih menguntungkan jika berjauhan. Karena penjelasan sterik bertentangan dengan hasil eksperimen, aturan Bent kemungkinan memainkan peran utama dalam penentuan struktur.
JCH Coupling constants
Mungkin pengukuran yang paling langsung dari karakter s dalam orbital pengikatan antara hidrogen dan karbon adalah melalui konstanta kopling 1H–13C yang ditentukan dari spektra NMR. Teori memprediksi bahwa nilai JCH berkorelasi dengan karakter s.[24] Secara khusus, konstanta kopling satu-ikatan 13C–1H, yaitu 1J13C-1H, berhubungan dengan karakter s fraksional dari orbital hibrida karbon yang digunakan untuk membentuk ikatan melalui hubungan empiris , di mana adalah karakter s. (Sebagai contoh, orbital atom hibrida sp3 murni dalam ikatan C–H metana memiliki 25% karakter s, menghasilkan konstanta kopling 500 Hz × 0,25 = 125 Hz, yang sangat sesuai dengan nilai eksperimental.)


| Molekul | JCH (dari proton metil) |
|---|---|
 Methane |
125 Hz |
 Acetaldehyde |
127 Hz |
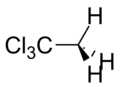 1,1,1–Trichloroethane |
134 Hz |
 Methanol |
141 Hz |
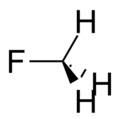 Fluoromethane |
149 Hz |
Seiring meningkatnya elektronegativitas substituen, jumlah karakter p yang diarahkan ke substituen juga meningkat. Hal ini menyisakan lebih banyak karakter s dalam ikatan ke proton metil, yang menyebabkan peningkatan konstanta kopling JCH.
Efek induktif
Efek induktif dapat dijelaskan dengan menggunakan kaidah Bent.[25] Efek induktif adalah transmisi muatan melalui ikatan kovalen dan kaidah Bent memberikan mekanisme untuk hasil tersebut melalui perbedaan hibridisasi. Dalam tabel di bawah,[26] ketika gugus-gugus yang terikat pada karbon pusat menjadi lebih elektronegatif, karbon pusat menjadi lebih menarik elektron sebagaimana diukur oleh konstanta substituen polar. Konstanta substituen polar serupa dengan nilai σ dari persamaan Hammett, karena peningkatan nilai menunjukkan kemampuan penarikan elektron yang lebih besar. Kaidah Bent menyatakan bahwa ketika elektronegativitas gugus meningkat, lebih banyak karakter p dialihkan ke arah gugus tersebut, yang menyisakan lebih banyak karakter s dalam ikatan antara karbon pusat dan gugus R. Karena orbital s memiliki kerapatan elektron yang lebih besar dekat inti daripada orbital p, kerapatan elektron dalam ikatan C–R akan lebih bergeser menuju karbon seiring meningkatnya karakter s. Hal ini membuat karbon pusat lebih menarik elektron terhadap gugus R.[10] Dengan demikian, kemampuan penarikan elektron dari substituen telah ditransfer ke karbon yang bersebelahan, sebagaimana diprediksi oleh efek induktif.
| Substituen | Konstanta substituen polar (nilai lebih besar menunjukkan kemampuan penarikan elektron lebih besar) |
|---|---|
 t–Butyl |
−0.30 |
 Methyl |
0.00 |
 Chloromethyl |
1.05 |
 Dichloromethyl |
1.94 |
 Trichloromethyl |
2.65 |
Teori formal
Kaidah Bent memberikan tingkat ketepatan tambahan pada teori ikatan valensi. Teori ikatan valensi mengusulkan bahwa ikatan kovalen terdiri atas dua elektron yang berada dalam orbital atomik yang saling bertumpang tindih, biasanya terhibridisasi, dari dua atom pengikatan. Asumsi bahwa sebuah ikatan kovalen merupakan kombinasi linear dari orbital atomik hanya dari dua atom pengikatan adalah sebuah pendekatan (lihat teori orbital molekul), tetapi teori ikatan valensi cukup akurat sehingga telah dan terus memiliki dampak besar pada cara ikatan dipahami.[1]
Dalam teori ikatan valensi, dua atom masing-masing menyumbangkan sebuah orbital atomik dan elektron dalam tumpang tindih orbital tersebut membentuk ikatan kovalen. Atom biasanya tidak menyumbangkan orbital mirip hidrogen murni untuk ikatan.[7] Jika atom hanya dapat menyumbangkan orbital mirip hidrogen, maka struktur tetrahedral metana yang telah dikonfirmasi secara eksperimental tidak mungkin terjadi karena orbital 2s dan 2p karbon tidak memiliki geometri tersebut. Kontradiksi itu dan kontradiksi lainnya mengarah pada pengusulan hibridisasi orbital. Dalam kerangka tersebut, orbital atomik diizinkan bercampur untuk menghasilkan jumlah orbital yang sama dengan bentuk dan energi yang berbeda. Dalam kasus metana, orbital 2s dan tiga orbital 2p karbon terhibridisasi untuk menghasilkan empat orbital sp3 ekuivalen, yang menyelesaikan ketidaksesuaian struktur tersebut. Hibridisasi orbital memungkinkan teori ikatan valensi menjelaskan geometri dan sifat dari sejumlah besar molekul secara sukses.
Dalam teori hibridisasi tradisional, orbital hibrida semuanya ekuivalen.[12][27] Yakni orbital atomik s dan p digabungkan untuk memberikan empat spi3 = 1⁄√4(s + √3pi) orbital, tiga spi2 = 1⁄√3(s + √2pi) orbital, atau dua spi = 1⁄√2(s + pi) orbital. Kombinasi ini dipilih untuk memenuhi dua syarat. Pertama, jumlah total kontribusi orbital s dan p harus ekuivalen sebelum dan sesudah hibridisasi. Kedua, orbital hibrida harus ortogonal satu sama lain.[27][28] Jika dua orbital hibrida tidak ortogonal, secara definisi mereka memiliki tumpang tindih orbital yang tidak nol. Elektron dalam orbital tersebut akan saling berinteraksi dan jika salah satu orbital itu terlibat dalam ikatan kovalen, orbital lainnya juga akan memiliki interaksi tak nol dengan ikatan tersebut, melanggar prinsip dua elektron per ikatan dalam teori ikatan valensi.
Untuk membangun orbital s dan p hibrida, biarkan orbital hibrida pertama diberikan oleh s + √λipi, di mana pi diarahkan menuju gugus pengikatan dan λi menentukan jumlah karakter p dalam orbital hibrida tersebut. Ini adalah jumlah berbobot dari fungsi gelombang. Sekarang pilih orbital hibrida kedua s + √λjpj, di mana pj diarahkan ke suatu arah dan λj adalah besarnya karakter p dalam orbital kedua ini. Nilai λj dan arah pj harus ditentukan sehingga orbital yang dihasilkan dapat dinormalisasi dan ortogonal terhadap orbital hibrida pertama. Orbital tersebut tentu dapat dinormalisasi karena merupakan jumlah dua fungsi gelombang ternormalisasi. Keortogonalan harus ditetapkan agar kedua orbital dapat digunakan dalam ikatan kovalen yang berbeda. Produk dalam dari orbital ortogonal harus nol dan menghitung produk dalam dari orbital yang dikonstruksi memberikan perhitungan berikut.
![{\displaystyle {\begin{aligned}\left\langle s+{\sqrt {\lambda _{i}}}p_{i}{\Big \vert }s+{\sqrt {\lambda _{j}}}p_{j}\right\rangle &=\langle s\mid s\rangle +{\sqrt {\lambda _{i}}}\langle s\mid p_{i}\rangle +{\sqrt {\lambda _{j}}}\langle s\mid p_{j}\rangle +{\sqrt {\lambda _{i}\lambda _{j}}}\langle p_{i}\mid p_{j}\rangle [4pt]&=1+0+0+{\sqrt {\lambda _{i}\lambda _{j}}}\cos \omega _{ij}=1+{\sqrt {\lambda _{i}\lambda _{j}}}\cos \omega _{ij}\end{aligned}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/550c4d0c3e888bf2515df5918559d3d42ca84e9e)
Orbital s ternormalisasi sehingga produk dalam ⟨ s | s ⟩ = 1. Selain itu, orbital s ortogonal terhadap orbital pi dan pj, yang menghasilkan dua suku bernilai nol. Terakhir, suku terakhir merupakan produk dalam dari dua fungsi ternormalisasi yang berada pada sudut ωij, yang menghasilkan cos ωij sesuai definisi. Namun, keortogonalan orbital pengikatan menuntut bahwa 1 + √λiλj cos ωij = 0, sehingga diperoleh teorema Coulson:[27][29]

Ini berarti bahwa empat orbital atomik s dan p dapat dihibridisasi ke arah apa pun selama semua koefisien λ memenuhi syarat di atas secara berpasangan untuk menjamin orbital yang dihasilkan ortogonal.
Kaidah Bent, bahwa atom pusat mengarahkan orbital dengan karakter p lebih besar ke substituen yang lebih elektronegatif, dapat diterapkan dengan mudah pada uraian di atas dengan mencatat bahwa peningkatan koefisien λi meningkatkan karakter p dari hibrida s + √λipi. Dengan demikian, jika atom pusat A berikatan dengan dua gugus X dan Y dan Y lebih elektronegatif daripada X, maka A akan menghibridisasi sehingga λX < λY. Teknik teoretis dan komputasi yang lebih canggih di luar kaidah Bent diperlukan untuk memprediksi geometri molekul secara akurat dari prinsip pertama, tetapi kaidah Bent memberikan heuristik yang sangat baik dalam menjelaskan struktur molekul.
Henry Bent awalnya mengusulkan kaidahnya pada tahun 1960 berdasarkan landasan empiris, tetapi beberapa tahun kemudian aturan tersebut didukung oleh perhitungan orbital molekul oleh Russell Drago.[23]
Aplikasi Kaidah Bent
Kaidah Bent mampu mengarakterisasi geometri molekul dengan akurat.[11][5] Kaidah Bent menyediakan kerangka kerja yang andal dan kuat untuk memprediksi sudut ikatan molekul. Akurasi dan presisi kaidah Bent dalam memprediksi geometri molekul dunia nyata terus menunjukkan kredibilitasnya.[5][15] Di luar prediksi sudut ikatan, kaidah Bent memiliki beberapa aplikasi penting dan menjadi perhatian besar bagi para kimiawan.[11][5][14][21][30] Kaidah Bent dapat diterapkan untuk menganalisis interaksi ikatan dan sintesis molekul.
Kaidah Bent dapat digunakan untuk memprediksi produk mana yang lebih disukai dalam suatu sintesis organik berdasarkan material awal.[14][30] Wang dkk. mempertimbangkan bagaimana substituen memengaruhi kesetimbangan silabenzena dan menemukan bahwa kaidah Bent memainkan peran penting dalam hasil tersebut.[14] Studi yang dilakukan Wang dkk. menunjukkan bagaimana kaidah Bent dapat digunakan untuk memprediksi jalur suatu sintesis dan stabilitas produk.[14] Menunjukkan aplikasi serupa, Dubois dkk. dapat membenarkan beberapa temuan mereka menggunakan kaidah Bent ketika mereka menemukan suatu reaksi bersifat irreversibel.[30] Kedua studi ini menunjukkan bagaimana kaidah Bent dapat membantu kimia sintetik. Mengetahui geometri molekul secara akurat berkat kaidah Bent memungkinkan kimiawan sintetik memprediksi stabilitas relatif produk.[14][30] Selain itu, kaidah Bent dapat membantu kimiawan memilih bahan awal untuk mendorong reaksi menuju produk tertentu.[14] Dengan demikian, kaidah Bent memungkinkan kimiawan sintetik untuk memiliki kontrol lebih besar terhadap reaksi yang diminati.
Lihat pula
Referensi
- ^ a b Weinhold, F.; Landis, C. L. (2005), Valency and Bonding: A Natural Donor-Acceptor Perspective (Edisi 1st), Cambridge: Cambridge University Press, ISBN 978-0-521-83128-4
- ^ a b c d e f g Bent, H. A. (1961), "An appraisal of valence-bond structures and hybridization in compounds of the first-row elements", Chem. Rev., 61 (3): 275–311, doi:10.1021/cr60211a005
- ^ Foster, J. P.; Weinhold, F. (1980), "Natural hybrid orbitals", J. Am. Chem. Soc., 102 (24): 7211–7218, Bibcode:1980JAChS.102.7211F, doi:10.1021/ja00544a007
- ^ Alabugin, I. V.; Bresch, S.; Gomes, G. P. (2015). "Orbital Hybridization: a Key Electronic Factor in Control of Structure and Reactivity". J. Phys. Org. Chem. 28 (2): 147–162. doi:10.1002/poc.3382.
- ^ a b c d e f g h i j Jonas, V.; Boehme, C.; Frenking, G. (1996-01-01). [[1](https://pubs.acs.org/doi/10.1021/ic951397o) "Bent's Rule and the Structure of Transition Metal Compounds"]. Inorganic Chemistry (dalam bahasa Inggris). 35 (7): 2097–2099. doi:10.1021/ic951397o. ISSN 0020-1669.
- ^ Alabugin, I. V.; Bresch, S.; Manoharan, M. (2014). "Hybridization Trends for Main Group Elements and Expanding the Bent's Rule Beyond Carbon: More than Electronegativity". J. Phys. Chem. A. 118 (20): 3663–3677. Bibcode:2014JPCA..118.3663A. doi:10.1021/jp502472u. PMID 24773162.
- ^ a b Pauling, L. (1931), "The nature of the chemical bond. Application of results obtained from the quantum mechanics and from a theory of paramagnetic susceptibility to the structure of molecules", J. Am. Chem. Soc., 53 (4): 1367–1400, Bibcode:1931JAChS..53.1367P, doi:10.1021/ja01355a027
- ^ Slater, J. C. (1931), "Directed Valence in Polyatomic Molecules", Phys. Rev., 37 (5): 481–489, Bibcode:1931PhRv...37..481S, doi:10.1103/PhysRev.37.481
- ^ Coulson, C. A. (1961), Valence (Edisi 2nd), Oxford: Clarendon Press
- ^ a b c Walsh, A. D. (1947), "The properties of bonds involving carbon", Discuss. Faraday Soc., 2: 18–25, doi:10.1039/DF9470200018
- ^ a b c d e f Ball, D. W.; Key, J. A. (16 September 2014). ["Molecular Shapes and Polarity". Pemeliharaan CS1: Banyak nama: authors list (link)](https://opentextbc.ca/introductorychemistry/chapter/molecular-shapes-and-polarity/}})
- ^ a b c d e f g Esselman, Brian J.; Block, Stephen B. (2019-01-08). [[2](https://pubs.acs.org/doi/10.1021/acs.jchemed.8b00316) "VSEPR-Plus: Correct Molecular and Electronic Structures Can Lead to Better Student Conceptual Models"]. Journal of Chemical Education (dalam bahasa Inggris). 96 (1): 75–81. Bibcode:2019JChEd..96...75E. doi:10.1021/acs.jchemed.8b00316. ISSN 0021-9584.
- ^ a b Gillespie, R. J. (2008-07-01). [[3](https://www.sciencedirect.com/science/article/pii/S0010854507001476) "Fifty years of the VSEPR model"]. Coordination Chemistry Reviews. 252 (12): 1315–1327. doi:10.1016/j.ccr.2007.07.007. ISSN 0010-8545.
- ^ a b c d e f g Wang, Xuerui; Huang, Ying; An, Ke; Fan, Jinglan; Zhu, Jun (November 2014). [[4](https://linkinghub.elsevier.com/retrieve/pii/S0022328X14004021) "Theoretical study on the interconversion of silabenzenes and their monocyclic non-aromatic isomers via the [1,3]-substituent shift: Interplay of aromaticity and Bent's rule"]. Journal of Organometallic Chemistry (dalam bahasa Inggris). 770: 146–150. doi:10.1016/j.jorganchem.2014.08.018.
- ^ a b c Ghosh, Dulal C.; Bhattacharyya, Soma (January 2005). [[5](https://onlinelibrary.wiley.com/doi/10.1002/qua.20690) "Computation of quantum mechanical hybridization and dipole correlation of the electronic structure of the F 3 B–NH 3 supermolecule"]. International Journal of Quantum Chemistry (dalam bahasa Inggris). 105 (3): 270–279. Bibcode:2005IJQC..105..270G. doi:10.1002/qua.20690. ISSN 0020-7608.
- ^ Kimura, Katsumi; Kubo, Masaji (1959-01-01). "Structures of Dimethyl Ether and Methyl Alcohol". The Journal of Chemical Physics. 30 (1): 151–158. Bibcode:1959JChPh..30..151K. doi:10.1063/1.1729867. ISSN 0021-9606.
- ^ Venkateswarlu, Putcha; Gordy, Walter (1955-07-01). "Methyl Alcohol. II. Molecular Structure". The Journal of Chemical Physics. 23 (7): 1200–1202. Bibcode:1955JChPh..23.1200V. doi:10.1063/1.1742240. ISSN 0021-9606.
- ^ Hankins, D.; Moskowitz, J. W.; Stillinger, F. H. (1970-12-15). "Water Molecule Interactions". The Journal of Chemical Physics. 53 (12): 4544–4554. Bibcode:1970JChPh..53.4544H. doi:10.1063/1.1673986. ISSN 0021-9606.
- ^ Hilton, A. Ray; Jache, Albert W.; Beal, James B.; Henderson, William D.; Robinson, R. J. (1961-04-01). "Millimeter Wave Spectrum and Molecular Structure of Oxygen Difluoride". The Journal of Chemical Physics. 34 (4): 1137–1141. Bibcode:1961JChPh..34.1137H. doi:10.1063/1.1731711. ISSN 0021-9606.
- ^ a b [[6](https://chem.libretexts.org/Bookshelves/Inorganic_Chemistry/Introduction_to_Organometallic_Chemistry_%28Ghosh_and_Balakrishna%29/01%3A_Introduction/1.02%3A_VSEPR_Theory_and_its_Utility) "1.2: VSEPR Theory and its Utility"]. Chemistry LibreTexts (dalam bahasa Inggris). 2019-08-15. Diakses tanggal 2023-12-06.
- ^ a b Grabowski, Sławomir J. (2011-11-10). [[7](https://pubs.acs.org/doi/10.1021/jp205019s) "Halogen Bond and Its Counterparts: Bent's Rule Explains the Formation of Nonbonding Interactions"]. The Journal of Physical Chemistry A (dalam bahasa Inggris). 115 (44): 12340–12347. Bibcode:2011JPCA..11512340G. doi:10.1021/jp205019s. ISSN 1089-5639. PMID 21970363.
- ^ Weinhold, F.; Landis, Clark R. (2012). Discovering Chemistry with Natural Bond Orbitals. Hoboken, N.J.: Wiley. hlm. 67–68. ISBN 9781118119969.
- ^ a b c Huheey, James E. (1983). Inorganic Chemistry (Edisi 3rd). Harper & Row. hlm. 230. ISBN 0-06-042987-9.
- ^ H. Friebolin (2008). Basic One- and Two- Dimensional NMR Spectroscopy (Edisi 4). VCH. ISBN 978-3-527-31233-7.
- ^ Bent, H. A. (1960), "Distribution of atomic s character in molecules and its chemical implications", J. Chem. Educ., 37 (12): 616–624, Bibcode:1960JChEd..37..616B, doi:10.1021/ed037p616
- ^ Taft Jr., R. W. (1957), "Concerning the Electron—Withdrawing Power and Electronegativity of Groups", J. Chem. Phys., 26 (1): 93–96, Bibcode:1957JChPh..26...93T, doi:10.1063/1.1743270
- ^ a b c Coulson, C. A. (1961), Valence (Edisi 2nd), Oxford: Clarendon Press, hlm. 203–5 Non–equivalent hybrids
- ^ Zhong, Ronglin; Zhang, Min; Xu, Hongliang; Su, Zhongmin (2016). "Latent harmony in dicarbon between VB and MO theories through orthogonal hybridization of 3σ g and 2σ u". Chemical Science (dalam bahasa Inggris). 7 (2): 1028–1032. doi:10.1039/C5SC03437J. ISSN 2041-6520. PMC 5954846. PMID 29896370.
- ^ Pfennig, Brian W. (2015). [[8](https://books.google.com/books?id=irRNCAAAQBAJ&dq=%22Coulson%27s+theorem%22&pg=PT543) "10"]. Principles of Inorganic Chemistry. John Wiley and Sons. ISBN 9781118859018. Diakses tanggal 11 August 2023.
Equation (10.11) is also known as Coulson's theorem
- ^ a b c d Dubois, Maryne A. J.; Rojas, Juan J.; Sterling, Alistair J.; Broderick, Hannah C.; Smith, Milo A.; White, Andrew J. P.; Miller, Philip W.; Choi, Chulho; Mousseau, James J.; Duarte, Fernanda; Bull, James A. (2023-05-19). "Visible Light Photoredox-Catalyzed Decarboxylative Alkylation of 3-Aryl-Oxetanes and Azetidines via Benzylic Tertiary Radicals and Implications of Benzylic Radical Stability". The Journal of Organic Chemistry (dalam bahasa Inggris). 88 (10): 6476–6488. doi:10.1021/acs.joc.3c00083. ISSN 0022-3263. PMC 10204094. PMID 36868184.
Konten ini disalin dari wikipedia, mohon digunakan dengan bijak.

